Fenyloketonuria, znana również jako PKU, to wrodzona choroba metaboliczna, diagnozowana już na wczesnym etapie życia. Nieleczona może prowadzić do poważnych konsekwencji neurologicznych. Warto więc wiedzieć, czym dokładnie jest PKU i w jaki sposób przebiega diagnostyka tej choroby.
Czym jest PKU?
PKU, czyli fenyloketonuria, to wrodzone zaburzenie metaboliczne spowodowane mutacją w genie odpowiedzialnym za produkcję enzymu hydroksylazy fenyloalaninowej (PAH). Enzym ten odpowiada za rozkład aminokwasu fenyloalaniny obecnego m.in. w białkach zwierzęcych i roślinnych.
U osoby zdrowej fenyloalanina przekształcana jest w tyrozynę – związek niezbędny do produkcji hormonów i neuroprzekaźników, takich jak dopamina. U chorych na PKU ten proces nie zachodzi prawidłowo, co prowadzi do nagromadzenia fenyloalaniny we krwi i mózgu. Zbyt wysokie jej stężenie ma toksyczny wpływ na układ nerwowy, może powodować zaburzenia rozwoju intelektualnego, drgawki, problemy z mową i koncentracją czy zmiany skórne i zaburzenia pigmentacji. Szybkie wdrożenie leczenia w postaci odpowiedniej diety niskobiałkowej pozwala na normalny, zdrowy rozwój.
Jeśli chcesz dowiedzieć się więcej na temat fenyloketonurii oraz życia z PKU, zajrzyj na stronę: https://www.vitapku.pl/.
Jak wygląda diagnoza fenyloketonurii?
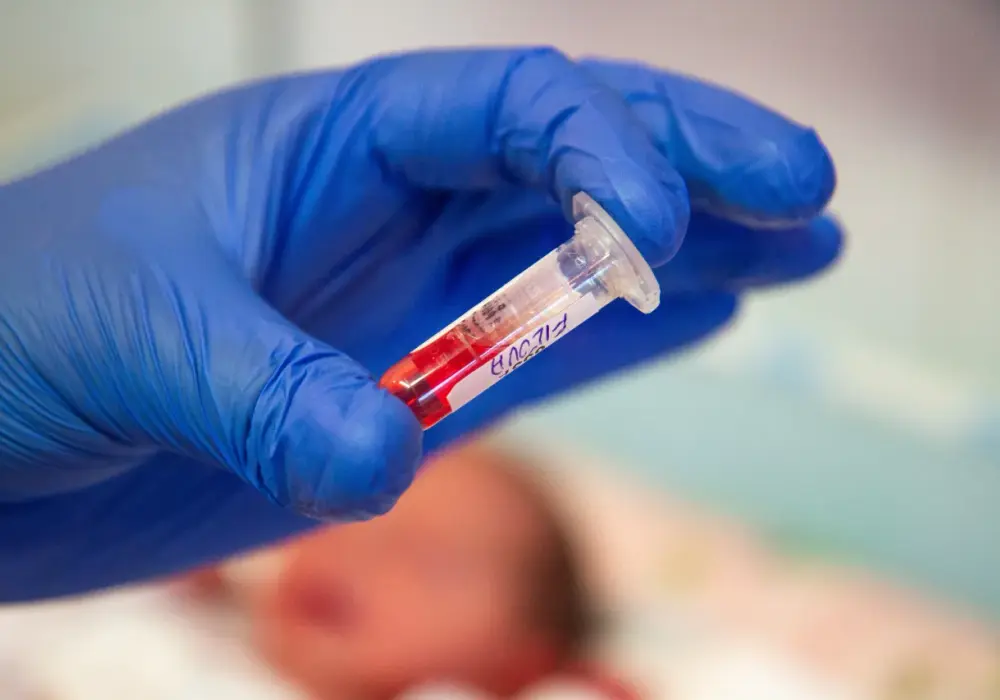
Wczesne rozpoznanie fenyloketonurii jest niezwykle istotne, dlatego w Polsce każde dziecko tuż po urodzeniu przechodzi obowiązkowe badania przesiewowe w kierunku PKU. Między 3. a 5. dobą życia, po rozpoczęciu karmienia, z pięty dziecka pobiera się kilka kropel krwi, a następnie nanosi ją na specjalną bibułę filtracyjną. Próbka trafia do laboratorium specjalistycznego, gdzie analizuje się stężenie fenyloalaniny we krwi. Dzięki temu można wykryć PKU jeszcze zanim pojawią się jakiekolwiek objawy.
W przypadku stwierdzenia nieprawidłowego wyniku rodzice są natychmiast informowani, a dziecko skierowane na badania potwierdzające, m.in. oznaczenie poziomu fenyloalaniny i tyrozyny w osoczu, testy enzymatyczne oraz badania genetyczne w celu identyfikacji mutacji w genie PAH. Potwierdzona diagnoza pozwala na niezwłoczne wdrożenie leczenia. Czas w tym przypadku jest na wagę złota – rozpoczęcie specjalistycznej diety w ciągu pierwszych 7-10 dni życia jest kluczowe dla uniknięcia nieodwracalnych uszkodzeń.
Dlaczego wczesne rozpoznanie PKU jest tak ważne?
Fenyloketonuria to choroba, którą można skutecznie kontrolować, ale tylko wtedy, gdy zostanie wcześnie wykryta. Brak odpowiedniego leczenia lub zbyt późna diagnoza mogą prowadzić do nieodwracalnych uszkodzeń mózgu. Szybka reakcja na pozytywny wynik testu przesiewowego i natychmiastowe wprowadzenie specjalistycznej diety eliminacyjnej, opartej na specjalnych produktach medycznych i kontrolowanym spożyciu białka, pozwala na utrzymanie poziomu fenyloalaniny w bezpiecznym zakresie. W rezultacie wcześnie zdiagnozowane i konsekwentnie leczone dzieci z PKU mogą rozwijać się prawidłowo, uczęszczać do szkoły, uprawiać sport i prowadzić pełne, zdrowe życie. Z tego względu programy badań przesiewowych noworodków uznaje się za jedno z największych osiągnięć współczesnej medycyny prewencyjnej.